WDL Tutorial
This tutorial is a way to get started with WDL if you are coming from snakemake or if you are use to a shared file system set up (ie. you can access and write out directories and files in a local file system). This is only possible in an uncontainerized case of using WDL. So we will start off with an uncontainerized version of the script and will move into a containerized version after. Ultimately, the design philosophy of the WDL language is to make it portable with cloud computing and containers. Due to a desire to make our scripts portable we will need to always assume that all our scripts outputs are ephemeral and lost forever unless it was an explicit File output.
For some background you can check out StaPH-B’s had their monthly meeting focused on WDL/Cromwell that was recorded on Feb 2020.
TL;DR
- This has all example wdl and config files to run wdl workflow with cromwell.
Make sure to also read the notes on versions of WDL!
Pre-Tutorial Installation
To be able to run this tutorial you will need to have installed cromwell, womtool, and wdltools. A method for install is found here.
Also, there is a couple more things that need to be installed. If you are using conda environments just install this in the same environment as before.
pip install biopython
pip install pandas
This wdl also requires a script from Kraken Tools that has a slight modification described here. This version of the script is found in the gitlab folder.
What is WDL?
The Workflow Description Language (WDL) is an open-source, community-driven data processing language that provides a way to specify data processing workflows with a human-readable and writeable syntax. WDL makes it straightforward to define complex analysis tasks, chain them together in workflows, and parallelize their execution. WDL was originally developed for the Broad Institute’s genomic analysis pipelines, but it has been widely adopted and developed by a global community. Read more about the WDL Community and how you can participate at the OpenWDL website.
There is a series of videos to get you started with wdl on youtube. Also, the Broad Institute has a page with tutorials and documentation.
There are pre-published wdl workflows on BioWDL and on Dockstore. You can create your own wdl script on Terra following the Broad Institute Tutorial. Or you can search Dockerstore and import a wdl into Terra following these instructions.
The Basics
A wdl script is broken down into:
- A workflow: the order tasks should be called in.
- inputs - are the input thats are needed for a particular command to run and you need to say what type of input you are expecting (ie. File, String, Float, Int). These will be the defined variables that are used in the
commandsection.- The types of variables that are allowed can be found here.
- You can add a
?behind a variable to make it an optional input.
- inputs - are the input thats are needed for a particular command to run and you need to say what type of input you are expecting (ie. File, String, Float, Int). These will be the defined variables that are used in the
- Tasks: tells what needs to be run for a particular task and contains a inputs, command(s), outputs and runtime.
- inputs - same as above.
- command - This is the command line you want executed and will contain the variables that were in the input using the syntax
${variable}. - output - The output you expect from your command. Again you need to say the type of the output to expect (ie. File etc.)
- runtime - These are the resources that you will need to execute your command. This is were you will add your docker image if you are using one.
- Note that not all runtime attributes are recognized depending on the backend you are using. See list here.
- meta - The metadata associated with the workflow. This is important because as its part of best practices. This information is parsed by Dockstore to allow for searching for wdls.
- parameter_meta - You can put and explanation for the expected inputs should be. See example here.

Snakemake comparisions
In snakemake terms:
tasksarerules- In wdl
commandswould be what you put in theshell:orrunportion of arulein snakemake. - In wdl
runtimewould be what you put in theresouresportion of arulein snakemake. I do thinks this clutters up the script a bit more than in a Snakefile, but you don’t have to have aconfig.jsonso pick your posion.
Versions of WDL
You can check the version of WDL that cromwell is reading by looking at the beginning of the cromwell output when you run your WDL. You should see something like this.
[2021-04-21 12:56:09,34] [info] MaterializeWorkflowDescriptorActor [dcf91504]: Parsing workflow as WDL draft-2
version 1.0) as the first line of your WDL file.You can find differences in the syntax by checking the version details on wdl’s github.
A major difference between the way this tutorial is written WDL version draft-2 and WDL version 1.0 is an input statement.
For example version draft-2:
version draft-2
import "task_qc.wdl" as qc
workflow read_QC {
input {
File read1_raw = "../Test_data/SRR11953697/SRR11953697_1.fastq"
File read2_raw = "../Test_data/SRR11953697/SRR11953697_2.fastq"
}
call qc.fastqc {
input:
read1 = read1_raw,
read2 = read2_raw,
read1_name = basename(read1_raw, '.fastq'),
read2_name = basename(read2_raw, '.fastq')
}
output {
File fastqc1_html = fastqc.fastqc1_html
File fastqc1_zip = fastqc.fastqc1_zip
File fastqc2_html = fastqc.fastqc2_html
File fastqc2_zip = fastqc.fastqc2_zip
}
}
The task_qc.wdl looks like this:
version draft-2
task fastqc {
input {
File read1
File read2
String read1_name
String read2_name
}
command {
fastqc --outdir $PWD ${read1} ${read2}
}
output {
File fastqc1_html = "${read1_name}_fastqc.html"
File fastqc1_zip = "${read1_name}_fastqc.zip"
File fastqc2_html = "${read2_name}_fastqc.html"
File fastqc2_zip = "${read2_name}_fastqc.zip"
}
runtime {
docker: "staphb/fastqc:0.11.8"
}
}
Running this workflow will cause some version of the error:
ERROR: Unexpected symbol (line 6, col 3) when parsing '_gen10'.
Expected rbrace, got input.
input {
^
input{} statement with version 1.0 and NOT draft-2. **version 1.0
import "task_qc.wdl" as qc
workflow read_QC {
input {
File read1_raw = "../Test_data/SRR11953697/SRR11953697_1.fastq"
File read2_raw = "../Test_data/SRR11953697/SRR11953697_2.fastq"
}
call qc.fastqc {
input:
read1 = read1_raw,
read2 = read2_raw,
read1_name = basename(read1_raw, '.fastq'),
read2_name = basename(read2_raw, '.fastq')
}
output {
File fastqc1_html = fastqc.fastqc1_html
File fastqc1_zip = fastqc.fastqc1_zip
File fastqc2_html = fastqc.fastqc2_html
File fastqc2_zip = fastqc.fastqc2_zip
}
}
Changing the version number in this case will fix the error here.
Our First WDL
Baby WDL
Our first baby wdl will be the traditional “Hello World”. The script will contain the following.
workflow baby_wdl {
call myTask
}
task myTask {
command {
echo "hello world"
}
output {
String out = read_string(stdout())
}
}
Now we run the wdl with cromwell.
Cromwell run Baby_wdl.wdl
This will just output “Hello World” in hot pink as part of cromwell’s output. The output here is just that we will have a string in the stdout. You can also just run without the output portion if you wanted. Nothing too special here.
Begin Tutorial
Now we can run through a similar tutorial as we did with Snakemake this will start out with running fastqc on a forward and reverse read for one sample. I ran this on Aspen using a conda environment following these instructions. Anaconda and Conda are explained here.
$PATH for security reasons so make sure you put the full path!!workflow tutorial {
call fastqc {
}
}
# Tasks #
task fastqc {
File fastq_R1 = "/$PATH/files/Sample_123_L001_R1.fastq"
File fastq_R2 = "/$PATH/files/Sample_123_L001_R2.fastq"
String base_name1 = sub(fastq_R1, '.fastq', '')
String base_name2 = sub(fastq_R2, '.fastq', '')
command {
mkdir -p "/scicomp/home-pure/qpk9/TOAST/fastqc"
module load fastqc/0.11.5
fastqc ${fastq_R1} -o "/$PATH/fastqc"
fastqc ${fastq_R2} -o "/$PATH/fastqc"
output {
File fastqc_zip_1 = "/$PATH/fastqc/${base_name1}_fastqc.html"
File fastqc_html_1 = "/$PATH/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "/$PATH/fastqc/${base_name2}_fastqc.html"
File fastqc_html_2 = "/$PATH/fastqc/${base_name2}_fastqc.html"
}
}
Here we can see that at the top there is a workflow statement, which will tell cromwell what order to run tasks in and that “calls” our tasks written below. Below the workflow is our task, which we start out by defining the type of the input files (here File or String) and where to find them. As we move toward a more samples we will be defining the input files in the workflow section and not in task as we have done here. Next, we have the actual commands as they would be run on the command line. Lastly, there is an output section that tells cromwell what to expect.
Notice we use the sub() function that allows us to replace “.fastq” with nothing so we have the base of our sample name. There is also a function basename(), but this only trims the extention you give (like this basename(name, ".fastq")) it so its less flexible than sub().
Further information on the base structure of a wdl workflow can be found here.
While this works, we need a way to make this work for more samples. So we will next make the script more general.
$PATH for security reasons so make sure you put the full path!!workflow converted_smake {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
call fastqc {
input:
fastq_R1 = fastq_R1,
fastq_R2 = fastq_R2,
base_name1 = base_name1,
base_name2 = base_name2
}
}
# Tasks #
task fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
command {
mkdir -p "/$PATH/fastqc"
module load fastqc/0.11.5
fastqc ${fastq_R1} -o "/$PATH/fastqc"
fastqc ${fastq_R2} -o "/$PATH/fastqc"
}
output {
File fastqc_zip_1 = "/$PATH/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "/$PATH/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "/$PATH/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "/$PATH/fastqc/${base_name2}_fastqc.html"
}
}
Note we have now added an input section in our call of the fastqc task in the workflow.
We can make sure that our wdl works before passing it off to cromwell by using the womtool validate like this:
womtool validate Converted_SM.wdl
It will either tell you the error or tell you Success!…
Now that we have the script we will use wdltools to create a template json for us where we will add in the path for the samples we want to run through the workflow.
wdltool inputs Converted_SM.wdl > convert_sm.inputs.json
The json that was created will have this format.
{
"<workflow name>.<task name>.<variable name>": "<variable type>"
}
Which looks like this:
{
"converted_smake.fastq_R1": "File",
"converted_smake.base_name2": "String",
"converted_smake.fastq_R2": "File",
"converted_smake.base_name1": "String"
}
Open this file in a text editor and add in the location for the files of interest. Make sure you provide FULL PATH to fastq files!
$PATH for security reasons so make sure you put the full path!!{
"converted_smake.fastq_R1": "/$PATH/files/Sample_123_L001_R1.fastq",
"converted_smake.base_name2": "Sample_123_L001_R2",
"converted_smake.fastq_R2": "/$PATH/files/Sample_123_L001_R2.fastq",
"converted_smake.base_name1": "Sample_123_L001_R1"
}
Note don’t put a comma after your last input here or cromwell will complain about it. Saying something like this…
Unexpected character '}' at input index 541 (line 9, position 1), expected '"':
}
^
Now we can run the workflow with these input files.
cromwell run Converted_SM.wdl --inputs convert_sm.inputs.json
Building a Workflow
I like to make workflows that only require changing a working directory and allowing it to build the file structure for me. So the first step will be that I change our json file.
$PATH for security reasons so make sure you put the full path!!{
"converted_smake.workdir" : "/$PATH/Tutorial",
"converted_smake.fastq_R1": "Sample_123_L001_R1.fastq",
"converted_smake.fastq_R2": "Sample_123_L001_R2.fastq"
}
Next we edit our wdl script:
workflow converted_smake {
File fastq_R1
File fastq_R2
String workdir
call fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + fastq_R1,
fastq_R2 = workdir + "/files/" + fastq_R2,
base_name1 = sub(fastq_R1, '.fastq', ''),
base_name2 = sub(fastq_R2, '.fastq', ''),
fastqcdir = workdir + "/fastqc"
}
}
task fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
Here we are able to have a workdir variable that allows us to concatenate strings with +.
Increasing Throughput - Scatter/Gather
Scatter
Now that we have a workflow up and running we will want to get it running on multiple samples, because no one has just one sample. We can use different input types to increase the number of samples that are put through the workflow.
Input types:
- Arrays, maps and pairs can be used as inputs. In python terms these are lists, dictionaries and tuples respectively.
- Arrays:
- Inside a workflow an array is referenced as
Array[File]orArray[String]orArray[Int]. - Arrays inside of a json look like this:
{ "tutorial.workdir" : "/$PATH/Tutorial", "tutorial.fastq_files": ["Sample_123_L001_R2.fastq","Sample_123_L001_R1.fastq","Sample_456_L001_R2.fastq","Sample_456_L001_R1.fastq"] }Here we will use an
Arraythat contains pairs so our json now looks like this:
- Inside a workflow an array is referenced as
- Arrays:
{
"tutorial.workdir" : "/scicomp/home-pure/qpk9/TOAST/Tutorial",
"tutorial.fastq_files_paired": [
{"left": "Sample_123_L001_R1.fastq", "right": "Sample_123_L001_R2.fastq"},
{"left": "Sample_456_L001_R1.fastq", "right": "Sample_456_L001_R2.fastq"} ]
}
The syntax we use in the workflow is Array[Pair[File, File]]
So our workflow changes to this:
workflow converted_smake {
String workdir
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = sub(sample.left, '.fastq', ''),
base_name2 = sub(sample.right, '.fastq', ''),
fastqcdir = workdir + "/fastqc"
}
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc $fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
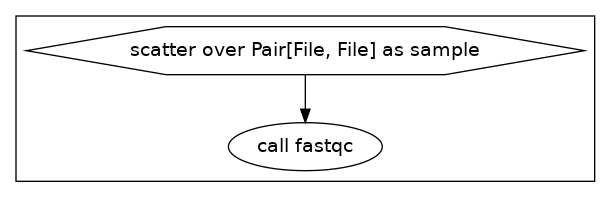
Here we state that we don’t just have a file, but an array of paired files with Array[Pair[File, File]] fastq_files_paired. We will also use scatter (sample in fastq_files_paired) {} to loop through each sample.
Our DAG from this workflow looks like this:
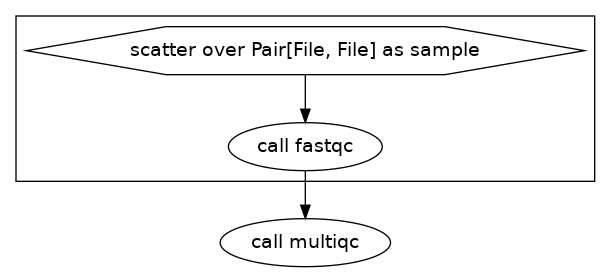
Gather
Next, we will want to gather the output of the task fastqc in an array (which is automatic) and run this output into another task multiqc.
workflow converted_smake {
String workdir
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = sub(sample.left, '.fastq', ''),
base_name2 = sub(sample.right, '.fastq', ''),
fastqcdir = workdir + "/fastqc"
}
}
call Multiqc {
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
workdir = workdir,
fastqcdir = workdir + "/fastqc",
multiqcdir = workdir + "/fastqc"
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
task Multiqc {
Array[File] fastqc_files_1
Array[File] fastqc_files_2
String workdir
String fastqcdir
String multiqcdir
command {
module load MultiQC/1.9
cd ${multiqcdir}
multiqc . --outdir ${multiqcdir}
}
output {
File multiqc_report = "${workdir}" + "/fastqc/multiqc_report.html"
}
}
We did not need to change our input json for this. We added a new call multiqc outside of the scatter statement. We then referenced the output of the task fastqc like this:
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
Lastly, we added the lines at the beginning of the task multiqc to state that the input is an array of files like this: Array[File] fastqc_files_1.
Now the DAG looks like this:
Parallelizing Commands
Now that we have fastqc running we might also want to run another program that takes the same inputs at the same time without waiting for the first job to finish.
Our diagram looks something like this:
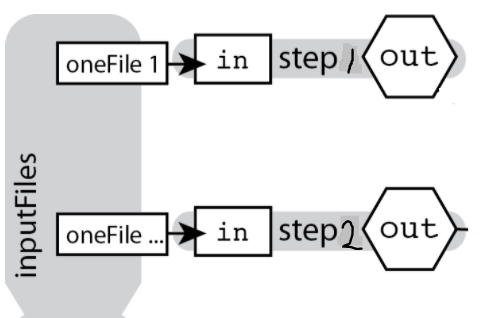
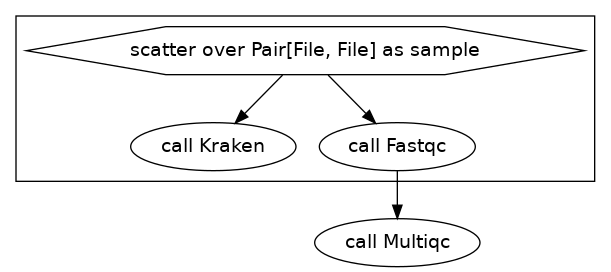
Here we will run fastqc and kraken at the same time.
workflow converted_smake {
String workdir
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = sub(sample.left, ".fastq", ""),
base_name2 = sub(sample.right, ".fastq", ""),
fastqcdir = workdir + "/fastqc"
}
call Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
krakendir = workdir + "/Kraken"
}
}
call Multiqc {
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
workdir = workdir,
fastqcdir = workdir + "/fastqc",
multiqcdir = workdir + "/fastqc"
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
task Multiqc {
Array[File] fastqc_files_1
Array[File] fastqc_files_2
String workdir
String fastqcdir
String multiqcdir
command {
module load MultiQC/1.9
cd ${multiqcdir}
multiqc . --outdir ${multiqcdir}
}
output {
File multiqc_report = "${workdir}" + "/fastqc/multiqc_report.html"
}
}
task Kraken {
File fastq_R1
File fastq_R2
String base_name
String krakendir
command {
mkdir -p ${krakendir}
module load kraken/2.0.8
kraken2 --use-names --threads 10 --db /$PATH/Kraken_DB/Updated_DB/ --report ${krakendir}/${base_name}_report.txt --paired ${fastq_R1} ${fastq_R2} --output ${krakendir}/${base_name.kraken
}
output {
File kraken = "${krakendir}" + "/${base_name}.kraken"
File report = "${krakendir}" + "/${base_name}_report.txt"
}
}
We added the task Kraken within the scatter{} to allow all samples to be sent through this process. Again we didn’t need to change our input file for this!
The DAG for this is:
Chaining Commands Together – Linear
Now we will take the output of our kraken command and use it as input for a python script that will pull out our sequnces of interest.
Our DAG will now be this:
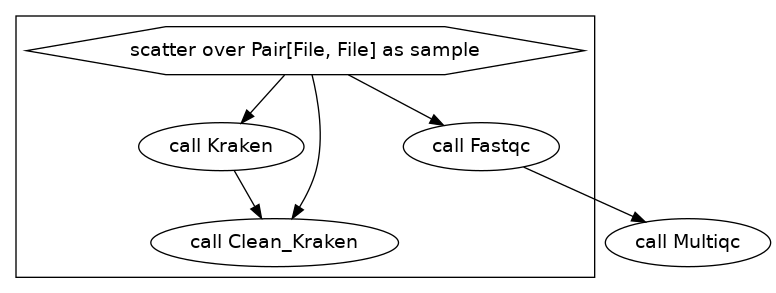
We did not need to change our input.json file, but our workflow now looks like this:
workflow converted_smake {
String workdir
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = sub(sample.left, ".fastq", ""),
base_name2 = sub(sample.right, ".fastq", ""),
fastqcdir = workdir + "/fastqc"
}
call Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
krakendir = workdir + "/Kraken"
}
call Clean_Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
clean_kraken_dir = workdir + "/Clean_Kraken",
kraken = Kraken.kraken,
report = Kraken.report
}
}
call Multiqc {
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
workdir = workdir,
fastqcdir = workdir + "/fastqc",
multiqcdir = workdir + "/fastqc"
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir} + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir} + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir} + "/fastqc/${base_name2}_fastqc.html"
}
}
task Multiqc {
Array[File] fastqc_files_1
Array[File] fastqc_files_2
String workdir
String fastqcdir
String multiqcdir
command {
module load MultiQC/1.9
cd ${multiqcdir}
multiqc . --outdir ${multiqcdir}
}
output {
File multiqc_report = "${workdir}" + "/fastqc/multiqc_report.html"
}
}
task Kraken {
File fastq_R1
File fastq_R2
String base_name
String krakendir
command {
mkdir -p ${krakendir}
module load kraken/2.0.8
kraken2 --use-names --threads 10 --db /$PATH/Kraken_DB/Updated_DB/ --report ${krakendir}/${base_name}_report.txt --paired ${fastq_R1} ${fastq_R2} --output ${krakendir}/${base_name}kraken
}
output {
File kraken = "${krakendir}" + "/${base_name}.kraken"
File report = "${krakendir}" + "/${base_name}_report.txt"
}
}
task Clean_Kraken {
File fastq_R1
File fastq_R2
String base_name
String clean_kraken_dir
File kraken
File report
command {
mkdir -p ${clean_kraken_dir}
python /$PATH/bin/KrakenTools-master/extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s ${fastq_R1} -s2 ${fastq_R2} -o ${clean_kraken_dir}/${base_name}_Kclean_R1.fastq -o2 ${clean_kraken_dir}/${base_name}_Kclean_R2.fastq --report ${report} -k ${kraken}
}
output {
File k_file_1 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R1.fastq"
File k_file_2 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R2.fastq"
}
}
The “big” difference here is we is that we link the input of clean_kraken from the task Kraken in the using the syntax kraken = Kraken.kraken, report = Kraken.report in the workflow portion like we did with the multiqc task.
Other Pipeline Patterns
Other pipeline patters are addressed here.
Aliasing and Subworkflows
This allows you to:
- Break up wdl scripts that get too long
- Alias a task to call it multiple times in the same script
- Import a common task into different wdl scripts (ie. QC used for most things) – This is called “subworkflows”
- Follow the convention of having tasks in a separate file from workflows and using import statements to call tasks into workflows.
To alias a task:
workflow converted_smake {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
call fastqc as round1 {
input:
fastq_R1 = fastq_R1,
fastq_R2 = fastq_R2,
base_name1 = base_name1,
base_name2 = base_name2
}
call fastqc as round2 {
input:
fastq_R1 = fastq_R1,
fastq_R2 = fastq_R2,
base_name1 = base_name1,
base_name2 = base_name2
}
}
To import a task as into a new wdl:
import "Converted_SM.wdl" as Converted_SM
workflow converted_smake_new {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
call Converted_SM.fastqc
call Converted_SM.fastqc as fastqc2
}
Notice here if you call twice you only need to alias the second time otherwise cromwell will tell you that you have duplicate outputs.
Running Inline Code
Python:
We will now add a new task at the beginning of our workflow that contains raw python in the command section. It’s just a silly little script that counts the number of reads in a fastq file and then returns a file named read_lengths.csv. Our DAG looks like this:
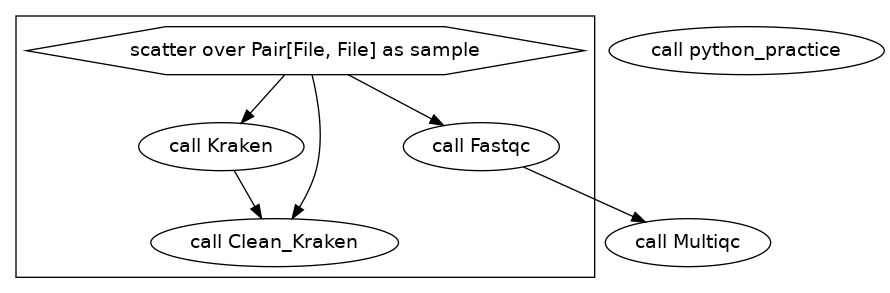
To do this, we are adding an input type called Array. Within the workflow we state that we will have an Array of files like this Array[File]. Our input json now looks like this:
Arrays just look like a python list.
$PATH for security reasons so make sure you put the full path!!{
"converted_smake.workdir" : "/$PATH/Tutorial",
"converted_smake.fastq_files": ["/$PATH/files/Sample_123_L001_R2.fastq","/$PATH/files/Sample_123_L001_R1.fastq","/$PATH/files/Sample_456_L001_R2.fastq","/$PATH/files/Sample_456_L001_R1.fastq"],
"converted_smake.fastq_files_paired": [
{left": "Sample_123_L001_R1.fastq", "right": "Sample_123_L001_R2.fastq"},
{left": "Sample_456_L001_R1.fastq", "right": "Sample_456_L001_R2.fastq"}
]
}
To have raw python within a command you use the following format:
command<<<
python <<CODE
import some_package
bla bla
if more bla
then do some bla
end of bla
CODE
>>>
<<< >>> notation and so you will need to use ~{variable} instead of ${variable} if you use the latter is that it gets a little too confusing to the parser about what’s bash and what’s WDL..Indent after python and then follow normal indention rules of python. Notice here we don’t have the { and instead have a series of <. Also, we need to have our imports to be part of the python code block.
If you want to run the same script, but not inline you can do this. Note, that while there was no arguments needed for the script I put in a couple as an example. This examples that var and arg2 are both string inputs.
task python_practice2 {
String var
String arg2
command <<<
python get_read_lengths.py --arg1=~{var} ~{arg2}
>>>
}
The wdl script now looks like this:
workflow converted_smake {
String workdir
Array[File] fastq_files
Array[Pair[File, File]] fastq_files_paired
call python_practice {
input:
workdir = workdir,
filesdir = workdir + "/files",
fastq_files = fastq_files
}
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = basename(sample.left, ".fastq"),
base_name2 = basename(sample.right, ".fastq"),
fastqcdir = workdir + "/fastqc"
}
call Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
krakendir = workdir + "/Kraken"
}
call Clean_Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
clean_kraken_dir = workdir + "/Clean_Kraken",
kraken = Kraken.kraken,
report = Kraken.report
}
}
call Multiqc {
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
workdir = workdir,
fastqcdir = workdir + "/fastqc",
multiqcdir = workdir + "/fastqc"
}
}
task python_practice {
Array[File] fastq_files
String filesdir
String workdir
command<<<
python <<CODE
import pandas as pd
import glob
import re
fastq_files = glob.glob(${filesdir} + "*.fastq")
df = pd.DataFrame(columns=['SeqID', 'ave_seq_length', "num_seqs"]) # make a empty dataframe
for file in fastq_files:
print("Analyzing {}".format(file))
seqID = re.search("^[^_]*", file).group(0) # Capture the Sequence ID at the beginning
num_seqs = 0
lengths = []
with open(file, "r") as f:
lines = f.readlines()
for line in lines:
line = line.strip('\n')
if line.startswith(('A', "G", "C", "T")):
num_seqs = num_seqs + 1
lengths.append(len(line))
else:
pass
ave_seq_length = int(sum(lengths)/len(lengths))
new_row = {SeqID': seqID, 'ave_seq_length': ave_seq_length, "num_seqs": num_seqs}
df = df.append(new_row, ignore_index=True)
df.to_csv(${workdir} + 'read_lengths.csv', sep=',', index=False)
CODE
>>>
output {
File read_lengths = "${workdir}" + "/read_lengths.csv"
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
task Multiqc {
Array[File] fastqc_files_1
Array[File] fastqc_files_2
String workdir
String fastqcdir
String multiqcdir
command {
module load MultiQC/1.9
cd ${multiqcdir}
multiqc . --outdir ${multiqcdir}
}
output {
File multiqc_report = "${workdir}" + "/fastqc/multiqc_report.html"
}
}
task Kraken {
File fastq_R1
File fastq_R2
String base_name
String krakendir
command {
mkdir -p ${krakendir}
module load kraken/2.0.8
kraken2 --use-names --threads 10 --db /$PATH/Kraken_DB/Updated_DB/ --report ${krakendir}/${base_name}_report.txt --paired ${fastq_R1} ${fastq_R2} --output ${krakendir}/${base_name}.kraken
}
output {
File kraken = "${krakendir}" + "/${base_name}kraken"
File report = "${krakendir}" + "/${base_name}_report.txt"
}
}
task Clean_Kraken {
File fastq_R1
File fastq_R2
String base_name
String clean_kraken_dir
File kraken
File report
command {
mkdir -p ${clean_kraken_dir}
python /$PATH/bin/KrakenTools-master/extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s ${fastq_R1} -s2 ${fastq_R2} -o ${clean_kraken_dir}/${base_name}_Kclean_R1.fastq -o2 ${clean_kraken_dir}/${base_name}_Kclean_R2.fastq --report ${report} -k ${kraken}
}
output {
File k_file_1 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R1.fastq"
File k_file_2 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R2.fastq"
}
}
Bash:
Cool, we have figured out how to run our python scripts now we will run some bash for practice.
Here is our DAG for this workflow.
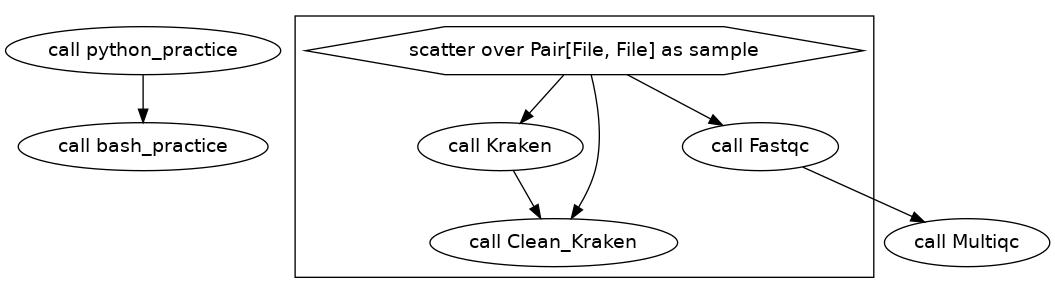
Next we will do a similar example with bash, in which we will open the read_lengths.csv file that was created and then extract the 3rd column using cut like this cat ${read_lengths} | cut -d, -f3.
The full workflow looks like this now:
# Workflow #
workflow converted_smake {
String workdir
Array[File] fastq_files
Array[Pair[File, File]] fastq_files_paired
call python_practice {
input:
workdir = workdir,
filesdir = workdir + "/files",
fastq_files = fastq_files
}
call bash_practice {input: read_lengths = python_practice.read_lengths}
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = basename(sample.left, ".fastq"),
base_name2 = basename(sample.right, ".fastq"),
fastqcdir = workdir + "/fastqc"
}
call Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
krakendir = workdir + "/Kraken"
}
call Clean_Kraken {
input:
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name = sub(sample.left, "_R1.fastq", ""),
clean_kraken_dir = workdir + "/Clean_Kraken",
kraken = Kraken.kraken,
report = Kraken.report
}
}
call Multiqc {
input:
fastqc_files_1 = Fastqc.fastqc_html_1,
fastqc_files_2 = Fastqc.fastqc_html_2,
workdir = workdir,
fastqcdir = workdir + "/fastqc",
multiqcdir = workdir + "/fastqc"
}
}
task python_practice {
Array[File] fastq_files
String filesdir
String workdir
command<<<
python <<CODE
import pandas as pd
import glob
import re
fastq_files = glob.glob(${filesdir} + "*.fastq")
df = pd.DataFrame(columns=['SeqID', 'ave_seq_length', "num_seqs"]) # make a empty dataframe
for file in fastq_files:
print("Analyzing {format(file))
seqID = re.search("^[^_]*", file).group(0) # Capture the Sequence ID at the beginning
num_seqs = 0
lengths = []
with open(file, "r") as f:
lines = f.readlines()
for line in lines:
line = line.strip('\n')
if line.startswith(('A', "G", "C", "T")):
num_seqs = num_seqs + 1
lengths.append(len(line))
else:
pass
ave_seq_length = int(sum(lengths)/len(lengths))
new_row = {SeqID': seqID, 'ave_seq_length': ave_seq_length, "num_seqs": num_seqs}
df = df.append(new_row, ignore_index=True)
df.to_csv(${workdir} + 'read_lengths.csv', sep=',', index=False)
CODE
>>>
output {
File read_lengths = "${workdir}" + "/read_lengths.csv"
}
}
task bash_practice{
File read_lengths
command<<<
cat ~{read_lengths} | cut -d, -f3
>>>
output {
String out = read_string(stdout())
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc/${base_name2}_fastqc.html"
}
}
task Multiqc {
Array[File] fastqc_files_1
Array[File] fastqc_files_2
String workdir
String fastqcdir
String multiqcdir
command {
module load MultiQC/1.9
cd ${multiqcdir}
multiqc . --outdir ${multiqcdir}
}
output {
File multiqc_report = "${workdir}" + "/fastqc/multiqc_report.html"
}
}
task Kraken {
File fastq_R1
File fastq_R2
String base_name
String krakendir
command {
mkdir -p ${krakendir}
module load kraken/2.0.8
kraken2 --use-names --threads 10 --db /$PATH/Kraken_DB/Updated_DB/ --report ${krakendir}/${base_name}_report.txt --paired ${fastq_R1} ${fastq_R2} --output ${krakendir}/${base_name}.kraken
}
output {
File kraken = "${krakendir}" + "/${base_name}.kraken"
File report = "${krakendir}" + "/${base_name}_report.txt"
}
}
task Clean_Kraken {
File fastq_R1
File fastq_R2
String base_name
String clean_kraken_dir
File kraken
File report
command {
mkdir -p ${clean_kraken_dir}
python /$PATH/bin/KrakenTools-master/extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s ${fastq_R1} -s2 ${fastq_R2} -o ${clean_kraken_dir}/${base_name}_Kclean_R1.fastq -o2 ${clean_kraken_dir}/${base_name}_Kclean_R2.fastq --report ${report} -k ${kraken}
}
output {
File k_file_1 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R1.fastq"
File k_file_2 = "${clean_kraken_dir}" + "/${base_name}_Kclean_R2.fastq"
}
}
The only big differences here are that we use the following format:
command<<<
code
>>>
Also, we use the output from the task python_practice by call bash_practice {input: read_lengths = python_practice.read_lengths} in the workflow.
The output of our task was written to the stdout file that is found at:
/$PATH/cromwell-executions/Tutorial/5b32bbae-ab73-46e7-99a9-e0f08475301e/call-bash_practice/execution/stdout
Running with Containers
Docker
Cromwell has documentation on how to add containers to your workflow.
Currently, StaPH-B has built many containers that are available on dockerhub, the source repository for these is on staph-B’s github page. You can find all of the containers on hub.docker.com by search for “staphb”.
wdl is configured to work with docker hub so we can swap out the module load portion of the tutorial and just pull in a docker container to run fastqc for us. This allows for reproducibility. Make sure you specify the version of fastqc if not then docker will just pull the latest one version of the container, but that will change so it won’t be reproducible then.
output. workflow tutorial {
String workdir
Array[File] fastq_files
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = basename(sample.left, ".fastq"),
base_name2 = basename(sample.right, ".fastq"),
}
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
command {
fastqc ${fastq_R1} -o .
fastqc ${fastq_R2} -o .
}
output {
File fastqc_zip_1 = "${base_name1}_fastqc.zip"
File fastqc_html_1 = "${base_name1}_fastqc.html"
File fastqc_zip_2 = "${base_name2}_fastqc.zip"
File fastqc_html_2 = "${base_name2}_fastqc.html"
}
runtime {
docker: "staphb/fastqc:0.11.8"
}
}
/$PATH/cromwell-executions/$workflow_name/1baab3f6-fca1-4eab-980a-78b33e9af11f/call-Fastqc/shard-0/execution/
and
/$PATH/cromwell-executions/$workflow_name/1baab3f6-fca1-4eab-980a-78b33e9af11f/call-Fastqc/shard-1/execution/
Here shard-0 and shard-1 hold the output of running fastqc on Sample_123_L001_R1.fastq and Sample_456_L001_R1.fastq, respectively.
Singularity
To run with Singularity rather than docker we will need to make a config file. Here is what is needed for Singularity:
# include statement
# this ensures defaults from application.conf
include required(classpath("application"))
backend {
# Override the default backend.
default: singularity
# The list of providers.
providers: {
#Singularity: a container safe for HPC
singularity {
# The backend custom configuration.
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory"
config {
run-in-background = true
runtime-attributes = """
String? docker
"""
submit-docker = """
singularity exec --bind ${cwd}:${docker_cwd} docker://${docker} ${job_shell} ${script}
"""
}
}
}
}
You can add more than one “provider” so if you need to add info for SGE submission you can. The Broad Institute has examples of config files for other providers. Documention on Singularity and cromwell is found here.
Also, because of how the data is mounted with singularity we can keep our previous version of the our wdl in which directories are created for us.
docker argument is keeped in the runtime as you would with running docker.workflow tutorial {
String workdir
Array[File] fastq_files
Array[Pair[File, File]] fastq_files_paired
scatter (sample in fastq_files_paired) {
call Fastqc {
input:
workdir = workdir,
fastq_R1 = workdir + "/files/" + sample.left,
fastq_R2 = workdir + "/files/" + sample.right,
base_name1 = basename(sample.left, ".fastq"),
base_name2 = basename(sample.right, ".fastq"),
fastqcdir = workdir + "/fastqc_container"
}
}
}
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
echo "Where is this going" > ${workdir}/PLEASEWORK1.txt
echo "Where is this going" > PLEASEWORK2.txt
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc_container/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc_container/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc_container/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc_container/${base_name2}_fastqc.html"
}
runtime {
docker: "staphb/fastqc:0.11.8"
}
}
docker argument is keeped in the runtime as you would with running docker.Running Cromwell on Different Backends
HPCs – SGE - No Containers
To run cromwell on Aspen and have it submit jobs to the cluster first we will create a config file singularity.conf.
# include statement
# this ensures defaults from application.conf
include required(classpath("application"))
backend {
default = SGE
providers {
SGE {
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory"
config {
concurrent-job-limit = 100
runtime-attributes = """
Int cpu = 1
Float? memory_gb
String? sge_queue
String? sge_project
"""
submit = """
qsub \
-terse \
-V \
-b y \
-N ${job_name} \
-wd ${cwd} \
-o ${out} \
-e ${err} \
-pe smp ${cpu} \
${"-l mem_free=" + memory_gb + "g"} \
${"-q " + sge_queue} \
${"-P " + sge_project} \
/usr/bin/env bash ${script}
"""
job-id-regex = "(\\d+)"
kill = "qdel ${job_id}"
check-alive = "qstat -j ${job_id}"
}
}
}
}
Now pass it to cromwell (non conda environment version):
java -Dconfig.file=singularity.conf -jar /scicomp/home-pure/qpk9/bin/cromwell-59.jar run Tutorial.wdl -i Tutorial.inputs.json
The default runtime parameters (AKA resources) will look something like this in the cromwell output:
[2021-03-30 16:36:14,18] [info] DispatchedConfigAsyncJobExecutionActor [800901c8testing_SGE.myTask:NA:1]: executing: qsub \
-terse \
-V \
-b y \
-N cromwell_800901c8_myTask \
-wd /$PATH/cromwell-executions/testing_SGE/800901c8-fd24-4429-acc9-3ecb46f7c51c/call-myTask \
-o /$PATH/cromwell-executions/testing_SGE/800901c8-fd24-4429-acc9-3ecb46f7c51c/call-myTask/execution/stdout \
-e /$PATH/cromwell-executions/testing_SGE/800901c8-fd24-4429-acc9-3ecb46f7c51c/call-myTask/execution/stderr \
-pe smp 1 \
-l mem_free=10.0g \
\
\
/usr/bin/env bash /$PATH/cromwell-executions/testing_SGE/800901c8-fd24-4429-acc9-3ecb46f7c51c/call-myTask/execution/script
[2021-03-30 16:36:14,81] [info] DispatchedConfigAsyncJobExecutionActor [800901c8testing_SGE.myTask:NA:1]: job id: 9000049
[2021-03-30 16:36:14,82] [info] DispatchedConfigAsyncJobExecutionActor [800901c8testing_SGE.myTask:NA:1]: Cromwell will watch for an rc file but will *not* double-check whether this job is actually alive (unless Cromwell restarts)
We can change this by adding runtime parameters to our task:
task Fastqc {
File fastq_R1
File fastq_R2
String base_name1
String base_name2
String workdir
String fastqcdir
command {
mkdir -p ${fastqcdir}
module load fastqc/0.11.5
fastqc ${fastq_R1} -o ${fastqcdir}
fastqc ${fastq_R2} -o ${fastqcdir}
}
output {
File fastqc_zip_1 = "${workdir}" + "/fastqc_container/${base_name1}_fastqc.zip"
File fastqc_html_1 = "${workdir}" + "/fastqc_container/${base_name1}_fastqc.html"
File fastqc_zip_2 = "${workdir}" + "/fastqc_container/${base_name2}_fastqc.zip"
File fastqc_html_2 = "${workdir}" + "/fastqc_container/${base_name2}_fastqc.html"
}
runtime {
memory: "5 GB"
cpu: "2"
}
}
Here we changed the standard 10GB of memory and 1cpu to 5GB and 2cpus. Now when we run cromwell we can see there is a change:
[2021-03-30 16:39:17,27] [info] DispatchedConfigAsyncJobExecutionActor [b1745a9dtesting_SGE.myTask:NA:1]: executing: qsub \
-terse \
-V \
-b y \
-N cromwell_b1745a9d_myTask \
-wd /$PATH/cromwell-executions/testing_SGE/b1745a9d-7b22-45db-bb02-7e75c2ed0b44/call-myTask \
-o /$PATH/cromwell-executions/testing_SGE/b1745a9d-7b22-45db-bb02-7e75c2ed0b44/call-myTask/execution/stdout \
-e /$PATH/cromwell-executions/testing_SGE/b1745a9d-7b22-45db-bb02-7e75c2ed0b44/call-myTask/execution/stderr \
-pe smp 2 \
-l mem_free=5.0g \
\
\
/usr/bin/env bash /$PATH/cromwell-executions/testing_SGE/b1745a9d-7b22-45db-bb02-7e75c2ed0b44/call-myTask/execution/script
[2021-03-30 16:39:17,86] [info] DispatchedConfigAsyncJobExecutionActor [b1745a9dtesting_SGE.myTask:NA:1]: job id: 9000050
[2021-03-30 16:39:17,87] [info] DispatchedConfigAsyncJobExecutionActor [b1745a9dtesting_SGE.myTask:NA:1]: Cromwell will watch for an rc file but will *not* double-check whether this job is actually alive (unless Cromwell restarts)
Other possible runtime parameters are found here.
HPCs – SGE - Using Singularity
To run cromwell on Aspen and have it submit jobs to the cluster first we will create a config file singularity_SGE.conf.
# include statement this ensures defaults from application.conf
include required(classpath("application"))
backend {
default = SGE
providers {
SGE {
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory"
config {
concurrent-job-limit = 100
runtime-attributes = """
Int cpu = 1
Float? memory_gb
String? sge_queue
String? sge_project
String? docker
"""
submit = """
qsub \
-terse \
-V \
-b y \
-N ${job_name} \
-wd ${cwd} \
-o ${out} \
-e ${err} \
-pe smp ${cpu} \
${"-l mem_free=" + memory_gb + "g"} \
${"-q " + sge_queue} \
${"-P " + sge_project} \
/usr/bin/env bash ${script}
"""
submit-docker = """
# Make sure the SINGULARITY_CACHEDIR variable is set. If not use a default
# based on the users home.
if [ -z $SINGULARITY_CACHEDIR ];
then CACHE_DIR=$HOME/.singularity/cache
else CACHE_DIR=$SINGULARITY_CACHEDIR
fi
# Make sure cache dir exists so lock file can be created by flock
mkdir -p $CACHE_DIR
LOCK_FILE=$CACHE_DIR/singularity_pull_flock
# Create an exclusive filelock with flock. --verbose is useful for
# for debugging, as is the echo command. These show up in `stdout.submit`.
flock --verbose --exclusive --timeout 900 $LOCK_FILE \
singularity exec --containall docker://${docker} \
echo "successfully pulled ${docker}!"
# Submit the script to SGE
qsub \
-terse \
-V \
-b y \
-N ${job_name} \
-wd ${cwd} \
-o ${cwd}/execution/stdout \
-e ${cwd}/execution/stderr \
-pe smp ${cpu} \
${"-l mem_free=" + memory_gb + "g"} \
${"-q " + sge_queue} \
${"-P " + sge_project} \
singularity exec --containall --bind ${cwd}:${docker_cwd} $IMAGE ${job_shell} ${docker_script}
"""
job-id-regex = "(\\d+)"
kill = "qdel ${job_id}"
check-alive = "qstat -j ${job_id}"
}
}
}
}
Then you pass the config file like this:
java -jar -Dconfig.file=singularity_SGE.conf ~/bin/cromwell-59.jar run Tutorial_Container.wdl --inputs Tutorial.inputs.json
Drawing a DAG
To visulaize what your workflow is doing you can use womtool and graphviz to build a diagram for you. First, use womtool to create a .dot file, then use graphviz (can be installed with conda) to convert the .dot file into a png image.
womtool graph Converted_SM.wdl > input.dot
dot -Tpng input.dot > output.png
Note on file paths
- When you are running wdl scripts locally you can either give relative paths or a google bucket path
gs://. - On Terra you use your workspace bucket or another google bucket, by putting the url in the attribute space in the workflows tab.
- Alternatively, on Terra you can create a “data model” under the data tab. Here you can import a tab del .tsv file that has an id and then the file location.
- Once the data table is in you can then go back to the workflows tab, select “Run workflow(s) with inputs defined by data table then select the id in the dropdown menu of step 1.
- You can find a video demonstrating this here.
- If you run
cromwell run --helpyou will see there is an argument--workflow-rootand you might (like me) think “EUREKA!” this will really get rid of all the annoying file paths in my wdl script. Alas, this isn’t the case :sob: see here.
Trouble shooting
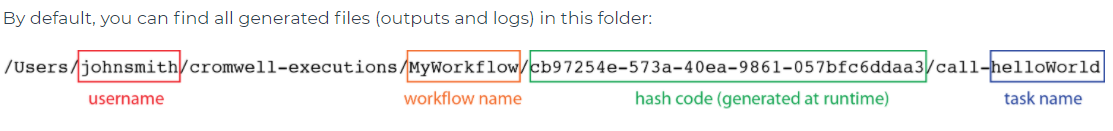
- If you are getting and error from cromwell and need to get the details, they are typically stored here:
$PATH/cromwell-executions/$worlflow_name/e35c4a3b-cecb-4956-8724-77ebafe03bfb/call-$task_name/execution/stderr
Note here that e35c4a3b-cecb-4956-8724-77ebafe03bfb is a random folder that you can either get from the output of cromwell or just the most recent one in that folder. You also need to add your workflow name and the task name.
- If your error is something like this:
Unable to build WOM node for WdlTaskCall 'Fastqc': No input base_name1 found evaluating inputs for expression base_name1 in (fastq_R2, base_name, kraken, fastq_R1, report)
Make sure that the variables that use use in the task in the error are also defined in the workflow portion.
- Specific to running on an HPC:
JobId are listed in the cromwell output like this:
BackgroundConfigAsyncJobExecutionActor [58bf62adconverted_smake.Fastqc:NA:1]: job id: 4391
BackgroundConfigAsyncJobExecutionActor [58bf62adconverted_smake.Kraken:NA:1]: job id: 4389
You can use a grep to get these lines out to check if memory or cpu requirements are a problem using qacct -j ## in theory, but I have found the numbers don’t match up correctly with the job IDs in Aspen.
Comparisons to Snakemake
- Cromwell will just give you an error that it failed, but doesn’t give you .err/.out file like snakemake can.
- Snakemake will only run files that need to be run so it doesn’t run everything link cromwell will do.
- Call caching can handle some of this, but you will need to edit this in the config file for cromwell. Also, there is some notes on that so becareful and read documentation first.
- There are also more details of call caching from Terra suport.