Snakemake Tutorial
Outline
Starting point: We have fastq files that have been run through kraken to identify taxonomy of reads.
The files that are the output of Kraken are found in the Kraken folder.
- Run our fastq files through fastqc.
- Filter our fastq files to retain only sequences that belong to Cryptosporidium.
- Rerun fastqc to see how removing “contamination” effect the quality of our files.
I like to use ATOM as my text editor when I write snakemake workflows since it provides out-of-the-box syntax highlighting for Snakemake.
How Snakemake Works
Snakemake documenation can be found at its readthedocs website.
The basics of snakemake:
- This is a rule based workflow management system.
- Rules generally specify:
- Inputs: Files that the rule operates on (i.e. dependencies)
- Outputs: Files that the rule creates
- An action: Some command to run. This can be either:
- BASH commands
- Python script (*.py)
- Inline python code
- R script (*.R)
- R markdown file (*.Rmd)
- Inputs and outputs are used by snakemake to determine the order for which rules are to be run. If a rule B has an input produced as an output of rule A, then rule B will be run after rule A.
- For determining whether output files have to be re-created, Snakemake checks whether the file modification date (i.e. the timestamp) of any input file of the same job is newer than the timestamp of the output file.
- This can be overridden by marking an input file with the
ancient()function. An example of ignoring timestamps is found here.
- This can be overridden by marking an input file with the
- All the arguments that can be used in a rule can be found here.
- There can be multiple inputs/outputs in rules
- Inputs/outputs can be named (using the = syntax), or just listed in order.
- These files can be referred to in the shell command (or python/R scripts)
- You can refer to the inputs/outputs of other rules like this:
rules.rule_name.output
- Rules generally specify:
- Snakemake will fill in wildcards based on what it finds in the output first. We will see an example of this later.
- For relative paths don’t use
./PATHjust leave it asPATH.
Running Snakemake
To run snakemake you will make a file called Snakefile,
rule fastqc:
input:
file = "files/Sample_123_L001_R1.fastq"
output:
"fastqc/Sample_123_L001_R1_fastqc.zip",
"fastqc/Sample_123_L001_R1_fastqc.html"
shell:
'''
mkdir -p fastqc
module load fastqc/0.11.5
fastqc files/Sample_123_L001_R1.fastq -o fastqc
'''
Note here that we are using a relative path based on where our Snakefile is at.
We can run snakemake by requesting the file that is wanted. For this to run you will also need to tell snakemake how many cores to use when running.
snakemake fastqc/Sample_123_L001_R1_fastqc.html --cores 1
Or just tell snakemake to run and it will go through the entire pipeline, which is only one rule right not so we get the same output.
snakemake --cores 1
You should see the output of fastqc at this point something like this…
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 fastqc
1
[Thu Apr 1 14:42:26 2021]
rule fastqc:
input: files/Sample_123_L001_R1.fastq
output: fastqc/Sample_123_R1_fastqc.zip, fastqc/Sample_123_L001_R1_fastqc.html
jobid: 0
Started analysis of Sample_123_L001_R1.fastq
Approx 5% complete for Sample_123_L001_R1.fastq
Approx 10% complete for Sample_123_L001_R1.fastq
Approx 15% complete for Sample_123_L001_R1.fastq
Approx 20% complete for Sample_123_L001_R1.fastq
Approx 25% complete for Sample_123_L001_R1.fastq
Approx 30% complete for Sample_123_L001_R1.fastq
Approx 35% complete for Sample_123_L001_R1.fastq
Approx 40% complete for Sample_123_L001_R1.fastq
Approx 45% complete for Sample_123_L001_R1.fastq
Approx 50% complete for Sample_123_L001_R1.fastq
Approx 55% complete for Sample_123_L001_R1.fastq
Approx 60% complete for Sample_123_L001_R1.fastq
Approx 65% complete for Sample_123_L001_R1.fastq
Approx 70% complete for Sample_123_L001_R1.fastq
Approx 75% complete for Sample_123_L001_R1.fastq
Approx 80% complete for Sample_123_L001_R1.fastq
Approx 85% complete for Sample_123_L001_R1.fastq
Approx 90% complete for Sample_123_L001_R1.fastq
Approx 95% complete for Sample_123_L001_R1.fastq
Analysis complete for Sample_123_L001_R1.fastq
[Thu Apr 1 14:43:30 2021]
Finished job 0.
1 of 1 steps (100%) done
If you already ran snakemake, snakemake will just tell you there is nothing to do and you will see this output:
Building DAG of jobs...
Nothing to be done.
Complete log: $PATH/.snakemake/log/2021-02-17T144540.917872.snakemake.log
Thanks snakemake! You can keep running this and nothing “bad” will happen, how refreshing…
We can update some of this code to make it a bit more universal. This will become more useful as we go on.
rule fastqc:
input:
file = "files/Sample_123_L001_R1.fastq"
params:
outdir = 'fastqc'
output:
"fastqc/Sample_123_L001_R1_fastqc.zip",
"fastqc/Sample_123_L001_R1_fastqc.html"
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
{input.file} is the same as {input[0]} if you didn’t use file= in the input parameter. Remember that python (and thus snakemake) is a 0 based language so the 0 element is the first element in the series.Scaling Our Pipeline with Wildcards
We can scale this pipeline by putting in wildcards that are placed in brackets rather than making one rule per sample.
rule fastqc:
input:
file = 'files/{sample}_R1.fastq'
params:
outdir = 'fastqc'
output:
"fastqc/{sample}_R1_fastqc.zip",
"fastqc/{sample}_R1_fastqc.html"
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
However, if we have run snakemake with snakemake fastqc/Sample_123_L001_R1_fastqc.html --cores 1 it will tell us there is nothing to be done because it fills in the wildcards based on what it finds in the output files which is only fastqc/Sample_123_L001_R1_fastqc.zip and fastqc/Sample_123_L001_R1_fastqc.html. However, if we delete these files and run snakemake with snakemake fastqc/Sample_123_L001_R1_fastqc.html --cores 1 we will get the following error.
WorkflowError:
Target rules may not contain wildcards. Please specify concrete files or a rule without wildcards.
This same error would have happened if you had ran snakemake --cores 1 because snakemake doesn’t know what file you want or if you want all of them. We can either just ask for only one file with snakemake fastqc/Sample_456_L001_R1_fastqc.zip --cores 1 for example, or create a new rule called rule all: that will allow us to tell snakemake that we want all the files (or whichever we want). The rule all will be put as the first rule in our snakefile by convention so we can keep track of it easier.
rule all:
input:
'fastqc/Sample_123_R1_fastqc.zip',
'fastqc/Sample_456_L001_R1_fastqc.zip',
'fastqc/Sample_123_R1_fastqc.html',
'fastqc/Sample_456_L001_R1_fastqc.html'
Cool, thats a bit better, but still a lot of typing and we don’t like that. We can use python syntax and the snakemake expand() function to make a list of file names we want.
SAMPLES = ['Sample_123_L001', 'Sample_456_L001']
rule all:
input:
expand('fastqc/{sample}_R1_fastqc.zip', sample=SAMPLES),
expand('fastqc/{sample}_R1_fastqc.html', sample=SAMPLES)
But again this is still a lot of typing. This can be cleaned up more by using snakemake’s glob_wildcards() to generate the file names for us. For this we give a path and file extention of files we want to collect the wildcard portion of the name from.
SAMPLES, = glob_wildcards('files/{sample}_R1.fastq')
rule all:
input:
expand('fastqc/{sample}_R1_fastqc.zip', sample=SAMPLES),
expand('fastqc/{sample}_R1_fastqc.html', sample=SAMPLES)
glob_wildcards() will act recursively, if the files are in the top directory. So I recommend you have your files in their own directory OR that you put in full paths for input and output files to avoid weird things happening…Whoops, this has only generated files for the R1 file and not the R2. We can add an additional wildcard to collect this information as well.
SAMPLES, READS, = glob_wildcards('files/{sample}_R{read}.fastq')
rule all:
input:
expand('fastqc/{sample}_R{read}_fastqc.zip', sample=SAMPLES, read=READS),
expand('fastqc/{sample}_R{read}_fastqc.html', sample=SAMPLES, read=READS)
rule fastqc:
input:
file = 'files/{sample}_R{read}.fastq'
params:
outdir = 'fastqc'
output:
"fastqc/{sample}_R{read}_fastqc.zip",
"fastqc/{sample}_R{read}_fastqc.html"
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
So now our dag is:
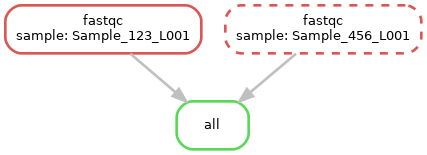 <figcaption></figcaption></figure>
<figcaption></figcaption></figure>
You could also explicity state what the wildcards will be instead:
SAMPLES, = glob_wildcards('{sample}_R1.fastq')
rule all:
input:
expand('fastqc/{sample}_R{read}_fastqc.zip', sample=SAMPLES, read=["1", "2"]),
expand('fastqc/{sample}_R{read}_fastqc.html', sample=SAMPLES, read=["1", "2"])
By default the expand function uses itertools.product to create every combination of the supplied wildcards. Expand takes an optional, second positional argument which can customize how wildcards are combined. How we currently wrote the expand function will give duplicates so we can add zip to clean this up.
We can use a simplified variant of expand(), called multiext(), that allows us to define a set of output or input files that just differ by their extension. This will clean things up a bit more.
Now our full baby snakemake file looks like this:
SAMPLES, READS, = glob_wildcards('files/{sample}_R{read}.fastq')
rule all:
input:
expand('fastqc/{sample}_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand('fastqc/{sample}_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
rule fastqc:
input:
file = '{sample}_R{read}.fastq'
params:
outdir = 'fastqc'
output:
multiext("fastqc/{sample}_R{read}_fastqc", ".zip", ".html")
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
Notes on Wildcards
-
Note: There is nothing special about
sampleyou could putsnakeorblablain the brackets and get the same result. That being said its usually good to make it a useful name, because someone else might need to read your code at some point. -
You can also contrain wildcards, which can be useful depending on what your file naming system is. As an example you add the following line to the top of the Snakefile. With this constraint, the wildcards can only contain letters, numbers, or underscores (not strictly true, but close enough most of the time).
wildcard_constraints:
sample = '\w+'
More details on wildcards can be found in the snakemake documenation.
input/output then you can’t have a non-wildcard output such as a commmon directory in the input/output. Put this in the params section. More information in the troubleshooting section below.Adding in Some Python
We have seen how to run programs that are already installed on the cluster, what about our own scripts? We can add python directly into the rule by swapping out the shell argument for run.
- A
shellaction executes a command-line instruction. - A
runaction executes Python code.
Here is a little script that opens a file and counts the number of sequences in the file and the average read length. A little silly, but you get the idea. We can add this to our Snakefile, but don’t forget to add the output to the rule all or snakemake won’t know you want to run this rule as part of the pipeline. This isn’t snakemake’s fault, just computers are dumb and will only do what you tell them even if it makes not sense.
run: rather than shell:.# import packages for python in rule python_practice
import pandas as pd
import glob
import re
rule python_practice:
input:
expand('files/{sample}_R{read}.fastq', zip, sample=SAMPLES, read=READS)
output:
'read_lengths.csv'
run:
fastq_files = glob.glob("files/*.fastq")
df = pd.DataFrame(columns=['SeqID', 'ave_seq_length', "num_seqs"]) # make a empty dataframe
for file in fastq_files:
print("Analyzing {}".format(file))
seqID = re.search("^[^_]*", file).group(0) # Capture the Sequence ID at the beginning
num_seqs = 0
lengths = []
with open(file, "r") as f:
lines = f.readlines()
for line in lines:
line = line.strip('\n')
if line.startswith(('A', "G", "C", "T")):
num_seqs = num_seqs + 1
lengths.append(len(line))
else:
pass
ave_seq_length = int(sum(lengths)/len(lengths))
new_row = {'SeqID': seqID, 'ave_seq_length': ave_seq_length, "num_seqs": num_seqs}
df = df.append(new_row, ignore_index=True)
df.to_csv('read_lengths.csv', sep=',', index=False)
Notice that you can write in python directly in the snakefile without it being part of a rule.
Running Python Scripts
If you have a python script you want to run this can be run as you would on the command line.
rule clean_kraken:
'''This rule will identify the sequences that belong to crypto and return fastq files with only these sequences'''
input:
file_1 = 'files/{sample}_R1.fastq',
file_2 = 'files/{sample}_R2.fastq',
kraken = 'Kraken/{sample}.kraken',
report = 'Kraken/{sample}_report.txt'
params:
outdir = 'Kraken_Cleaned'
output:
k_file_1 = 'Kraken_Cleaned/{sample}_Kclean_R1.fastq',
k_file_2 = 'Kraken_Cleaned/{sample}_Kclean_R2.fastq'
message: '''--- Running extract_kraken_reads.py to get fastq files without contamination. ---'''
shell:
'''
mkdir -p {params.outdir}
python extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s {input.file_1} -s2 {input.file_2} -o {output.k_file_1} -o2 {output.k_file_2} --report {input.report} -k {input.kraken}
'''
Notice here we added the message: argument. When executing snakemake, a short summary for each running rule is given to the console. This can be overridden by specifying a message for a rule as we did here.
Another way to run a python script (or any type of script) would be to use script: rather than shell: argument and have the python script make the output directory for us.
rule clean_kraken:
'''This rule will identify the sequences that belong to crypto and return fastq files with only these sequences'''
input:
file_1 = 'files/{sample}_R1.fastq',
file_2 = 'files/{sample}_R2.fastq',
kraken = 'Kraken/{sample}.kraken',
report = 'Kraken/{sample}_report.txt'
params:
outdir = 'Kraken_Cleaned'
output:
k_file_1 = 'Kraken_Cleaned/{sample}_Kclean_R1.fastq',
k_file_2 = 'Kraken_Cleaned/{sample}_Kclean_R2.fastq'
message: '''--- Running extract_kraken_reads.py to get fastq files without contamination. ---'''
script:
"extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s {input.file_1} -s2 {input.file_2} -o {output.k_file_1} -o2 {output.k_file_2} --report {input.report} -k {input.kraken}"
So currently our full Snakemake file looks like this:
#import packages for python in rule python_practice
import pandas as pd
import glob
import re
SAMPLES, READS, = glob_wildcards('files/{sample}_R{read}.fastq')
rule all:
input:
expand('fastqc/{sample}_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand('fastqc/{sample}_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
'read_lengths.csv',
expand('Kraken_Cleaned/{sample}_Kclean_R{read}.fastq', zip, sample=SAMPLES, read=READS)
rule fastqc:
input:
file = '{sample}_R{read}.fastq'
params:
outdir = 'fastqc'
output:
multiext("fastqc/{sample}_R{read}_fastqc", ".zip", ".html")
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
rule python_practice:
input:
expand('files/{sample}_R{read}.fastq', zip, sample=SAMPLES, read=READS)
output:
'read_lengths.csv'
run:
fastq_files = glob.glob("files/*.fastq")
df = pd.DataFrame(columns=['SeqID', 'ave_seq_length', "num_seqs"]) # make a empty dataframe
for file in fastq_files:
print("Analyzing {}".format(file))
seqID = re.search("^[^_]*", file).group(0) # Capture the Sequence ID at the beginning
num_seqs = 0
lengths = []
with open(file, "r") as f:
lines = f.readlines()
for line in lines:
line = line.strip('\n')
if line.startswith(('A', "G", "C", "T")):
num_seqs = num_seqs + 1
lengths.append(len(line))
else:
pass
ave_seq_length = int(sum(lengths)/len(lengths))
new_row = {'SeqID': seqID, 'ave_seq_length': ave_seq_length, "num_seqs": num_seqs}
df = df.append(new_row, ignore_index=True)
df.to_csv('read_lengths.csv', sep=',', index=False)
rule clean_kraken:
'''This rule will identify the sequences that belong to crypto and return fastq files with only these sequences'''
input:
file_1 = 'files/{sample}_R1.fastq',
file_2 = 'files/{sample}_R2.fastq',
kraken = 'Kraken/{sample}.kraken',
report = 'Kraken/{sample}_report.txt'
params:
outdir = 'Kraken_Cleaned'
output:
k_file_1 = 'Kraken_Cleaned/{sample}_Kclean_R1.fastq',
k_file_2 = 'Kraken_Cleaned/{sample}_Kclean_R2.fastq'
message: '''--- Running extract_kraken_reads.py to get fastq files without contamination. ---'''
shell:
'''
mkdir -p {params.outdir}
python extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s {input.file_1} -s2 {input.file_2} -o {output.k_file_1} -o2 {output.k_file_2} --report {input.report} -k {input.kraken}
'''
Your Turn
Now that you have a bit of experience with snakemake go ahead and write a new rule to run fastqc on the new files that have been created. When you have given it a shot click below to see my solution.
Answer
Your code should look something like this:
# import packages for python in rule python_practice
import pandas as pd
import glob
import re
SAMPLES, READS, = glob_wildcards('files/{sample}_R{read}.fastq')
rule all:
input:
expand('fastqc/{sample}_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand('fastqc/{sample}_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
expand('Kraken_Cleaned/{sample}_Kclean_R{read}.fastq', zip, sample=SAMPLES, read=READS),
expand('fastqc_Cleaned/{sample}_Kclean_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand('fastqc_Cleaned/{sample}_Kclean_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
"read_lengths.csv"
rule fastqc:
input:
file = 'files/{sample}_R{read}.fastq'
params:
outdir = 'fastqc'
output:
multiext("fastqc/{sample}_R{read}_fastqc", ".zip", ".html")
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
rule python_practice:
input:
expand('files/{sample}_R{read}.fastq', zip, sample=SAMPLES, read=READS)
output:
'read_lengths.csv'
run:
fastq_files = glob.glob("files/*.fastq")
df = pd.DataFrame(columns=['SeqID', 'ave_seq_length', "num_seqs"]) # make a empty dataframe
for file in fastq_files:
print("Analyzing {}".format(file))
seqID = re.search("^[^_]*", file).group(0) # Capture the Sequence ID at the beginning
num_seqs = 0
lengths = []
with open(file, "r") as f:
lines = f.readlines()
for line in lines:
line = line.strip('\n')
if line.startswith(('A', "G", "C", "T")):
num_seqs = num_seqs + 1
lengths.append(len(line))
else:
pass
ave_seq_length = int(sum(lengths)/len(lengths))
new_row = {'SeqID': seqID, 'ave_seq_length': ave_seq_length, "num_seqs": num_seqs}
df = df.append(new_row, ignore_index=True)
df.to_csv('read_lengths.csv', sep=',', index=False)
rule clean_kraken:
'''This rule will identify the sequences that belong to crypto and return fastq files with only these sequences'''
input:
file_1 = 'files/{sample}_R1.fastq',
file_2 = 'files/{sample}_R2.fastq',
kraken = 'Kraken/{sample}.kraken',
report = 'Kraken/{sample}_report.txt'
params:
outdir = 'Kraken_Cleaned'
output:
k_file_1 = 'Kraken_Cleaned/{sample}_Kclean_R1.fastq',
k_file_2 = 'Kraken_Cleaned/{sample}_Kclean_R2.fastq'
message: '''--- Running extract_kraken_reads.py to get fastq files without contamination. ---'''
shell:
'''
echo 'I am running on node:' `hostname`
mkdir -p {params.outdir}
python extract_kraken_reads.py --include-children --fastq-output --taxid 5806 -s {input.file_1} -s2 {input.file_2} -o {output.k_file_1} -o2 {output.k_file_2} --report {input.report} -k {input.kraken}
'''
rule fastqc_2:
input:
file = 'Kraken_Cleaned/{sample}_Kclean_R{read}.fastq'
params:
outdir = 'fastqc_Cleaned'
output:
multiext("fastqc_Cleaned/{sample}_Kclean_R{read}_fastqc", ".zip", ".html")
shell:
'''
mkdir -p {params.outdir}
module load fastqc/0.11.5
fastqc {input.file} -o {params.outdir}
'''
If this drop down doesn’t seem to work on the static webpage then you can go here.
Re-Running Rules
If you make a change and need to re-run a rule, there are a few options:
- If you modify any file that an output depends on, and then rerun snakemake, everything downstream from that file is re-run.
For example, if we modify an output (for example Sample_123_L001_R2.fastq) by running touch files/Sample_123_L001_R2.fastq at the command line this will update the time stamp on this file which then triggers a rerun of the fastqc and python_practice rules when snakemake --cores 1 is run.
- If you modify the code behind a rule, you can force the re-run of the rule by using the
-fflag
snakemake -f python_practice --cores 1 # forces this step to run
snakemake all --cores 1 # runs everything needed to get the files listed in the rule all:
- If you just want to re-run everything, you can use the
-Fflag. This forces a rule to run as well as every rule it depends on.
“Checkpoints”
There has been a time in which I want all my files to finish up to a certain rule before processing to the next rule. I don’t know if this is the best way to do this, but I have created a rule that requires all my files as in the input using expand() and had it create a blank file that the next rule downstream required as input:. I could forsee errors with this so be careful…
Dynamic/Checkpoints
Snakemake provides experimental support for dynamic files using the dynamic() function. Dynamic files can be used whenever one has a rule for which the number of output files is unknown before the rule was executed.
Here is a blog on checkpoints/dynamic files. Snakemakes documention also covers this.
Some Helpful Commands
There are lots of flags snakemake uses you can find a list here
To list all the rules in the file run one of the following.
snakemake --list
or
snakemake -l
To understand why each rule is being run use the --reason flag.
snakemake --reason
or
snakemake -r
To have snakemake go through a dry run of its workflow so you can make sure its doing what you want before you submit 100 jobs us the --dryrun flag. This will only show what would have been run without running any commands.
snakemake --dryrun
To visualize your workflow you can generate a .png image with:
snakemake --dag | dot -Tpng > dag.png
In this case the DAG looks like this:

It should be noted that dag drawing won’t like it if you have print statements directly in the Snakefile and not in a rule.
Keeping Things Tidy
If you have a lot of paths that might change you can simply your snakefile my creating a config.json file. For example, we can create one that has a working directory defined.
{
"workdir": "/scicomp/home-pure/USER_ID/Snakemake_Tutorial/"
}
This can then be called within your snakefile like this (using rule all as an expample):
configfile: '$PATH_TO_CONFIG/config.json'
rule all:
input:
expand(config['workdir'] + 'fastqc/{sample}_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand(config['workdir'] + 'fastqc/{sample}_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
expand(config['workdir'] + 'Kraken_Cleaned/{sample}_Kclean_R{read}.fastq', zip, sample=SAMPLES, read=READS),
expand(config['workdir'] + 'fastqc_Cleaned/{sample}_Kclean_R{read}_fastqc.zip', zip, sample=SAMPLES, read=READS),
expand(config['workdir'] + 'fastqc_Cleaned/{sample}_Kclean_R{read}_fastqc.html', zip, sample=SAMPLES, read=READS),
config['workdir'] + "read_lengths.csv"
Protected and Temporary Files
Output files can be marked as protected in the Snakefile and it will be ‘locked’ (write permissions removed) after creation so that it’s harder to accidentally delete it.
Alternately, you can mark a file as temp and it will be deleted as soon as any rules that depend on it are run. This is a good way to automatically remove intermediate files that take up lots of hard disk space.
rule example_rule:
input:
"some/file/input.txt",
output:
protected("my_protected_file.txt"),
temp("my_temporary_file.txt"),
script: "do_things.py"
Troubleshooting (AKA mistakes I made)
- Snakemake will get mad if you have directories in the output (with no wildcard in them) if there are wildcards in other output files. If a rule’s inputs have wildcards then it expects all outputs to have a wildcard as well. Otherwise it duplicates it and it will make directories multiple times.
Snakemake will give this error:
SyntaxError:
Not all output, log and benchmark files of rule xxx contain the same wildcards. This is crucial though, in order to avoid that two or more jobs write to the same file.
Solution: put directories under params: rather than output.
- If you get this error:
SyntaxError in line XX of /$PATH/Snakefile:
EOF in multi-line statement (Snakefile, line XX)
or
SyntaxError in line 307 of /$PATH/Snakefile:
invalid syntax
The most common cause is not having a comma after every line for your inputs and outputs.
- Snakemake will be annoyed if you have a non-assigned object after an assigned one in the input or output. Snakemake will complain with this error:
SyntaxError in line XXXX of /$PATH/Snakefile:
positional argument follows keyword argument
DON’T do this:
rule example:
input:
script = 'runme.sh',
examplefile.txt
DO this instead:
rule example:
input:
examplefile.txt,
script = 'runme.sh'
- If you see this error
Building DAG of jobs...
CyclicGraphException in line XXX of /$PATH/Snakefile:
Cyclic dependency on rule examplerule.
There is a matching file in your input and output arguments so doublecheck the rule that snakemake complains about. You can only have it in one location.
- Running ‘awk’ in ‘shell’ commnad of snakemake
If you have statements that contain {} in them then snakemake will complain about this as you pass arguments to snakemakes shell using {} so you will just need to escape the {} by using `` instead. Yay, an easy fix!